Why do short-lived lung infections lead to long-lasting lung damage?
Study points to mechanism of post-viral lung damage; suggests targets of intervention
 Kangyun Wu
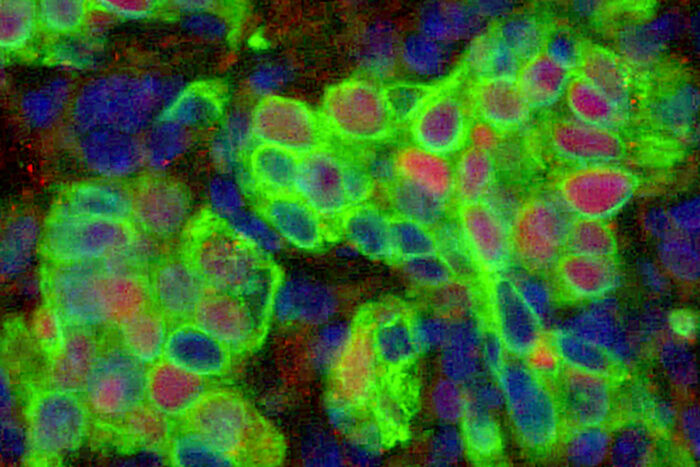
Kangyun WuBasal epithelial stem cells (green) containing the protein IL-33 (red) are visible in a sample of lung tissue (pictured). Researchers at Washington University School of Medicine in St. Louis have found that such stem cells multiply during and after a viral respiratory infection, sometimes triggering a detrimental remodeling process that can cause persistent lung disease long after the virus has been cleared.
The deadliest time in a viral respiratory illness sometimes is actually after the virus is cleared from the body. Destructive processes that are set in motion during an infection crest in the weeks after the virus is defeated, leading to organ damage that can cause chronic illness or even death. After an initial bout of COVID-19, for example, some people struggle with persistent cough, difficulty breathing and shortness of breath — signs of ongoing lung disease.
Researchers at Washington University School of Medicine in St. Louis have found clues to just how lung damage develops in the aftermath of a respiratory infection. Studying mice, they found that infection triggers the expression of a protein called IL-33, which is needed for stem cells in the lung to overgrow into air spaces, and increases mucus production and inflammation in the lung. The findings, published Aug. 24 in the Journal of Clinical Investigation, reveal potential points of intervention to prevent chronic lung damage caused by viral infections.
“Vaccines, antivirals, antibody therapies are all helpful, but they are not a solution for people who are already on the road to progressive disease,” said senior author Michael J. Holtzman, MD, the Selma and Herman Seldin Professor of Medicine and a professor of cell biology & physiology. “We’ve gotten better at taking care of the acute illness due to COVID-19, but what happens after that initial injury phase is still a major obstacle to a better outcome. At this point, we are also faced with tens of millions of people who already had infection, and a high percentage of them are having long-term disease, especially with respiratory symptoms. We don’t have a treatment that can correct the problem.”
It’s long been recognized that acute respiratory infections can lead to chronic lung disease. Children hospitalized with respiratory syncytial virus, for example, are two to four times more likely to develop asthma that persists for long periods, maybe even for a lifetime. How exactly an acute respiratory infection triggers chronic disease, however, is not fully understood, making it difficult to develop therapies to prevent or treat it.
As part of this study, Holtzman and colleagues, including first author Kangyun Wu, PhD, an instructor in medicine, studied mice infected with Sendai virus. Sendai doesn’t cause serious disease in people, but it naturally infects other animals including mice and causes respiratory infections that develop much like respiratory infections in people.
The researchers examined lung tissues from mice 12 and 21 days after infection with Sendai virus, and compared the samples to lung tissues of uninfected mice. They found that two populations of stem cells help maintain the barrier between the lung and the outside world in uninfected mice. After infection with Sendai virus, however, these two populations separately begin to multiply and spread into air spaces. Basal cells take over small airways and air sacs while AT2 cells remain confined to air sacs. Some of the new basal cells become mucus-producing cells while others release molecules that recruit immune cells to the lungs. Altogether, the process results in lungs with less air space, more mucus and ongoing inflammation that together interfere with breathing.
Further experiments showed that this process hinges on the protein IL-33. Under normal conditions, IL-33 increases in the nuclei of lung stem cells in response to stress or injury and helps the lung repair damaged barriers. During and after infection, though, IL-33 can take on a more detrimental role.
To assess the role of IL-33 in post-viral lung damage, the researchers genetically modified mice to lack IL-33 in the basal set of lung stem cells. The scientists then infected those mice — and a separate group of unmodified mice — with Sendai virus. The two groups of mice were equally effective at fighting off an initial Sendai virus infection. But three weeks after infection, the lungs of the mice that lacked IL-33 exhibited less cellular overgrowth, mucus and inflammation, indicating that they had fewer signs of harmful lung changes. At seven weeks after infection, the mice without IL-33 in basal cells also had higher oxygen levels in their blood and less airway hyperresponsiveness, both of which are signs of improvement in their chronic lung disease.
“These results were really nice to see because getting rid of IL-33 and in turn losing basal stem cells could have made things worse,” Holtzman said. “The engineered mice could have died because they were no longer able to perform the normal repair of the viral damage to the lung barrier. But that’s not the case. The mice lacking this population of basal cells instead had much better outcomes. That’s what we’re excited about. These findings put us on firm ground to find therapies that correct the bad behavior of basal stem cells.”
Targeting steps on the pathway between IL-33 and basal cell activation could form the basis of broadly effective therapies to prevent or treat lung disease caused by a variety of viruses and perhaps other forms of injury in the lung and other sites where the body meets the outside world, Holtzman said.
“The lung has a pretty stereotyped response to injury, including viral injury,” Holtzman said. “The specific type of virus, the genetics of the host, the severity of the initial illness — all of these things influence the outcome, but they’re just matters of degrees. You still see the same key elements across conditions, and that’s why we believe that there can be a common strategy for treatment. We have a drug discovery program to find such a common strategy, and this study fits well with that.”