Novel trial targets genetic form of ALS
Washington University School of Medicine researchers are investigating a novel targeted therapy to treat the neurodegenerative disorder ALS

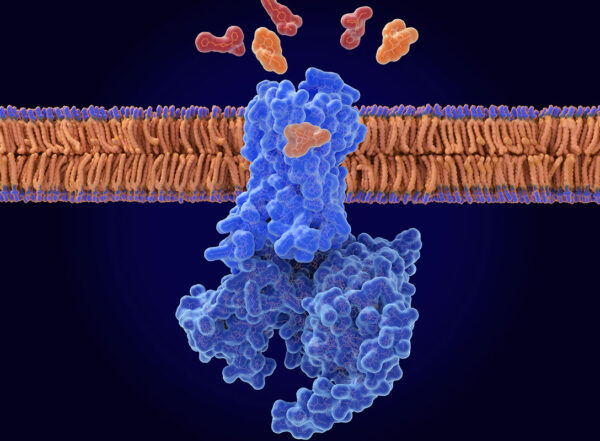
Researchers at Washington University are working on a new therapy that targets a gene affected in certain forms of ALS. Image courtesy of Jonathan Bailey, National Human Genome Research Institute (NHGRI).
A diagnosis of ALS, or amyotrophic lateral sclerosis, is devastating.
Once a patient is diagnosed with the neurodegenerative disease, also known as Lou Gehrig’s disease, average survival is just three to five years. But some patients with a particular genetic form of ALS may die within one year.
This form of the disease involves a mutation in the SOD1 gene, which researchers at Washington University are targeting. They have completed a phase 1 safety trial and are now moving on to phase 2 studies, with the ultimate goal of prolonging survival.
“This is a targeted therapy for one genetic form of the disease,” says neurologist and lead investigator Timothy Miller, MD, PhD. “This type of targeted approach is used in clinical trials for cancer, but it is new to do this for neurodegenerative diseases and ALS.”
Testing a gene-targeting molecule
More than 100 changes, or mutations, in the SOD1 gene account for 2 percent of all ALS cases. But the mutations’ exact role in causing ALS is unknown. Rather than try to understand that connection, Miller and his colleagues are focused on a therapy that blocks gene’s production of the SOD1 protein.
In the trials, Miller delivers a molecule called an antisense oligonucleotide into the cerebrospinal fluid (the fluid found in the brain and spine). This molecule blocks the SOD1 gene and has the goal of lowering SOD1protein concentrations there. The trials are the very first to deliver such a molecule directly into human cerebrospinal fluid to treat a widespread central nervous system disorder.
“We hope that this type of approach, which is targeted to the known problem, will be able to slow down the disease in a way that hasn’t been done before,” Miller says. “We don’t know what to expect in humans as the trials move forward, but many of us believe this is the best shot we have in slowing down the disease.”
After promising results in a rat model of the disease, Miller and colleagues—including co-lead investigator Merit Cudkowicz, MD, of Harvard—performed a phase 1 human trial in four groups of eight patients each. In each group, six patients received the antisense oligonucleotide molecule and two received a placebo.
Though many study participants had minor adverse events, such as back pain and nausea, there were no dose-limiting toxic effects and no safety or tolerability concerns related to the oligonucleotide. Patients were allowed to re-enroll in later groups, and the researchers found that re-enrollment and re-treatment also were well-tolerated.
“We were very encouraged by the trial results,” Miller says. “We found that the therapy was very safe. We’re enthusiastic about moving forward with phase 2 of the trial.”
The phase 1 results were published in The Lancet Neurology in March 2013.
Phase 2 will likely involve about 40 patients, and the primary goal will again be to use an antisense oligonucleotide to lower SOD1 protein in the cerebrospinal fluid. The researchers will also pay great attention to safety as the antisense oligonucleotide is administered at higher doses and for longer periods of time.
Strong family history
Miller explains that most patients with SOD1 ALS have a strong family history of ALS and therefore are tested for the gene. This genetic testing is important because anyone diagnosed with this form of the disease will be a potential candidate for the targeted therapy that Miller and his colleagues are studying.
Considering the fact that some patients with SOD1 ALS die nine to 12 months after their diagnosis, Miller says the researchers are moving as fast as they reasonably can to begin phase 2 of the trial.
“These kinds of trials of novel therapies take important commitment from the patient population,” Miller says. “It’s important to recognize the brave volunteers who step forward to participate and make these trials go—the families and the patients. This is not easy for them to do.”
Washington University was one of four clinical sites involved in the phase I trial. The others were Massachusetts General Hospital, Johns Hopkins and the Methodist Neurological Institute in Houston.
“We are mapping out a novel way to treat neurodegenerative syndromes,” Miller says. “This approach could be applied to other neurodegenerative diseases, including some forms of Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and other conditions. And one could develop the exact same approach for any new genetic target defined for ALS.”
To find out if this trial is still enrolling, please contact clinical research specialist Jennifer Jockel-Balsarotti at 314-362-8624 or balsarottij@neuro.wustl.edu.
Editors note: The trial is supported by the ALS Association, the Muscular Dystrophy Association and Isis Pharmaceuticals, maker of the antisense oligonucleotide used in the trial.