New strategy shows potential to block nerve loss in neurodegenerative diseases
Research into how key molecule triggers axon death may lead to new therapies
 Yurie Yamada
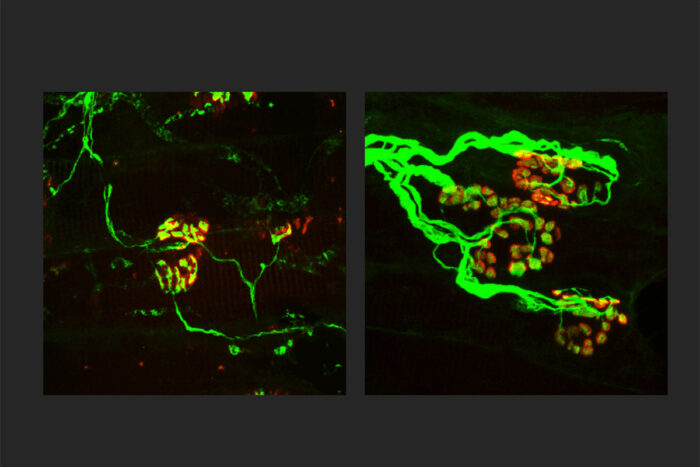
Yurie YamadaResearchers at Washington University School of Medicine in St. Louis are working toward a treatment for neurodegenerative diseases that targets SARM1, a key molecule in the death of axons, the wiring of the nervous system. Shown, the axons (green) are thinner in a rodent model of an inherited axonal peripheral neuropathy (left). When SARM1 is missing from rodents with the disorder, the axons are thicker (right) and indistinguishable from normal, healthy axons. Finding ways to block SARM1 could lead to new therapies for a broad range of neurodegenerative diseases.
Two new studies from Washington University School of Medicine in St. Louis support development of a broadly applicable treatment for neurodegenerative diseases that targets a molecule that serves as the central executioner in the death of axons, the wiring of the nervous system.
Blocking this molecular executioner prevents axon loss, which has been implicated in many neurodegenerative diseases, from peripheral neuropathies to Parkinson’s disease, and glaucoma to amyotrophic lateral sclerosis (ALS).
The new studies, both published Oct. 26 in the Journal of Clinical Investigation, reveal surprising details about how the molecule — called SARM1 — triggers axon death that underlies the development of neurodegenerative diseases. The research also points to new therapeutic approaches for diseases defined by axon loss.
“We desperately need treatments for neurodegenerative diseases,” said co-senior author Jeffrey Milbrandt, MD, PhD, the James S. McDonnell Professor and head of the Department of Genetics. “With the evidence of SARM1’s central role in these diseases, we’re very interested in finding ways to block this molecule — whether with small molecule inhibitors or gene therapy techniques. Our latest research suggests we also may be able to interfere with its ability to drive damaging neuroinflammation. We’re hopeful this work will lead to effective new therapies across a range of neurodegenerative and neuroinflammatory diseases.”
In 2017, Milbrandt and co-senior author Aaron DiAntonio, MD, PhD, the Alan A. and Edith L. Wolff Professor of Developmental Biology, discovered that SARM1 is an enzyme that can promote neurodegeneration. Soon after, they co-founded a startup company called Disarm Therapeutics to boost the development of drug compounds that inhibit SARM1 for the treatment of diseases characterized by axon degeneration. In 2020, Disarm Therapeutics was acquired by Eli Lilly and Company to further the development of SARM1-targeted therapies for neurodegenerative diseases.
In healthy neurons, SARM1 is always switched off. But after injury or due to disease, SARM1 becomes active. Activated SARM1 is an arsonist — burning so much cellular energy that the axons can’t survive. This energy crisis triggers axons to disintegrate.
To understand more about SARM1’s role in triggering axon destruction, the researchers studied a mysterious and extremely rare progressive neuropathy syndrome — so rare, it lacks a name. This rare disease turned out to be a good model for understanding the role of the immune system in neuroinflammatory conditions generally. Sequencing patient genomes, the researchers found that the axon loss was caused by genetic errors in the gene NMNAT2, whose normal function keeps SARM1 turned off. Due to these genetic errors, SARM1 is constantly activated, which triggers axon destruction. The researchers used the CRISPR gene-editing technique to reproduce these mutations in mice. Like people with the syndrome, these mice survived to adulthood but had worsening motor dysfunction, loss of peripheral axons and, importantly, an infiltration of immune cells called macrophages.
The researchers were surprised to find that reducing the number of macrophages reversed the axon loss and disease symptoms in the mice. The study suggests that SARM1 not only contributes directly to axon loss but also plays a role in driving neuroinflammation that only serves to compound the problems. The findings also suggest that some neurodegenerative conditions could be treated with immune modulating drugs that block macrophages or other inflammatory immune cells.
In the second paper, the researchers investigated the possible role of SARM1 in Charcot-Marie-Tooth disease type 2a, a common form of inherited peripheral neuropathy and a good model to study axon loss generally. Patients with this disease have progressive loss of motor and sensory axons and develop difficulty walking, muscle weakness, and tingling or burning sensations in the hands and feet. The disease is caused by a mutation in an important protein in mitochondria, the energy factories of cells. The mutation, in a protein called mitofusin2, impairs the normal function of mitochondria. Much research has focused on the abnormal mitochondria, assuming they must be the root of the problem in this disease.
Surprisingly, the researchers found that deleting SARM1 in a rodent model of Charcot-Marie-Tooth disease type 2a stopped most of the problems the animals exhibited — independent of the diseased mitochondria. Eliminating SARM1 blocked or slowed axon death, muscle atrophy, mitochondrial abnormalities and problems with neuromuscular junctions, where the neurons interface with muscle. Even with the mutant mitofusin2 protein present, deleting SARM1 protected the mitochondria from further degradation and dysfunction.
“When we block SARM1, we not only protect the axons, we get much healthier mitochondria,” DiAntonio said. “This was a complete surprise, but we are hopeful it could be relevant in many neurodegenerative diseases where mitochondrial damage is central, such as Parkinson’s disease, as many neurodegenerative diseases have a component of mitochondrial dysfunction.”