New clues found to understanding relapse in breast cancer
Tumor mutations found in patients point to progressive disease
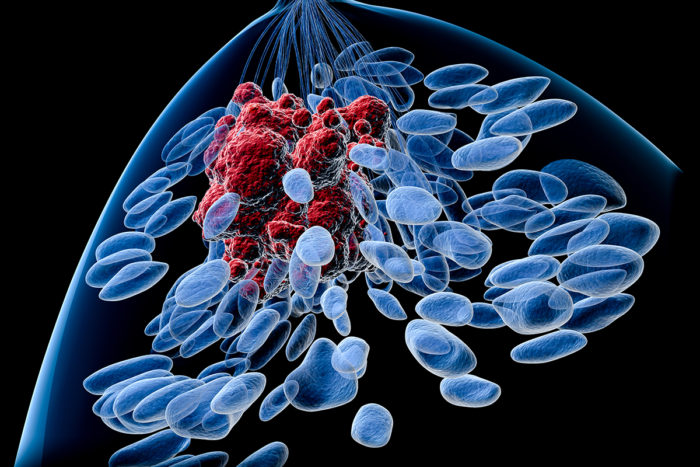 Getty Images
Getty ImagesShown is an artist's rendering of a breast tumor (red). Blue areas depict normal breast tissue. A new study identifies mutations associated with relapse in ER positive breast cancer — knowledge that could lead to better therapies.
A large genomic analysis has linked certain DNA mutations to a high risk of relapse in estrogen receptor positive breast cancer, while other mutations were associated with better outcomes, according to researchers from Washington University School of Medicine in St. Louis, the Baylor College of Medicine and the University of British Columbia.
The knowledge could help predict which patients are most likely to have their cancer return and spread, and could help guide treatment decisions. It also opens the door to developing more aggressive treatments for patients with the newly identified high-risk mutations.
The study appears Sept. 4 in the journal Nature Communications.
The researchers analyzed tumor samples from more than 2,500 patients with estrogen receptor positive breast cancer, one of the most common forms of the disease. These cancer cells have receptors that bind to the hormone estrogen in the nucleus of the cell and drive tumor growth.
More than 266,000 women in the United States are diagnosed with invasive breast cancer each year, according to the American Cancer Society, and about 70 percent of cases are estrogen receptor positive.
ER positive breast cancer patients have a number of treatment options that block the estrogen receptor to stop tumor growth. Such hormonal therapies are effective and less toxic than traditional chemotherapy and radiation. But some tumors develop resistance to these treatments, mutating in ways that fuel growth independent of the presence of estrogen. These types of mutations are of great interest because they are responsible for the majority of deaths due to breast cancer.
“We would like to help doctors identify patients who are likely to do well versus those who are likely to have a recurrence,” said first author Obi L. Griffith, PhD, an assistant professor of medicine and an assistant director of The McDonnell Genome Institute at Washington University School of Medicine. “Those with mutations that are associated with a good prognosis may need less intensive therapy than they might otherwise receive. But if a patient’s tumor has mutations linked to high risk of relapse, it’s useful to know that early so they can be treated with more aggressive therapies or even potential investigational therapies that could be targeted to their specific mutations.”
The new study confirmed past work showing that relatively common mutations in genes called MAP3K1 and TP53 had opposite effects on tumor aggressiveness. Patients with MAP3K1 mutations did well, while those with TP53 mutations were likely to have a recurrence. The study also identified three genes — DDR1, PIK3R1 and NF1 — with relatively uncommon mutations that were associated with cancer recurrence and spreading.
“Although mutations in DDR1 and NF1 are considered rare, they are associated with early relapse, which makes them much more common in patients who unfortunately die from the disease and, thus, could be critical therapeutic targets,” said Matthew J. Ellis, MB, BChir, PhD, of the Baylor College of Medicine. “Their identification also gives us very important molecular clues into the nature of aggressive tumor behavior.”
“It is likely that these genes will now be included in gene panel tests, particularly when clinical trials are developed that target these mutations,” Ellis added.
One unique element of this study was the age of the samples. Many were over 20 years old, allowing the researchers to know the full history of how the patients fared. But unlike more recent cancer sequencing studies, neither healthy tissue nor blood samples from those patients were collected or stored alongside the tumor. That means the old tumor samples couldn’t be compared with normal DNA from the same patients. Comparing the DNA of healthy tissue versus tumor tissue in the same patient can help identify mutations driving the cancer.
According to co-first author Nicholas C. Spies, a graduate student in Griffith’s lab, scientists now have enough healthy genomes sequenced to be able to compare, on a broad population level, normal genomes to cancer genomes and use big data bioinformatics methods to pull out the mutations likely to be driving cancer, even in old samples that can’t be directly compared with healthy DNA from the same patient.
The researchers also pointed out the importance of continuing to sequence cancer genomes.
“With all the breast cancer sequencing that’s been done, it’s tempting to think we’ve found everything of importance,” Griffith said. “But this study tells us there is still more to discover.”