Defect in debilitating neurodegenerative disease reversed in mouse nerves
Drug compound may help against Charcot-Marie-Tooth disease
 Matt Miller
Matt MillerGerald Dorn, PhD, (left) and staff scientist Agostinho Rocha, PhD, study mitochondria in relation to Charcot-Marie-Tooth disease in Dorn's lab. The researchers have developed a drug compound that shows promise as a future treatment for this disease.
Scientists have developed a new drug compound that shows promise as a future treatment for Charcot-Marie-Tooth disease, an inherited, often painful neurodegenerative condition that affects nerves in the hands, arms, feet and legs. The researchers used the compound to treat the nerves of mice harboring the genetic defects that cause the disease.
The new study, from Washington University School of Medicine in St. Louis, challenges some conventional wisdom regarding how patients with this disease lose the ability to move their limbs.
The study appears April 20 in the journal Science.
Charcot-Marie-Tooth is the most common inherited degenerative disease of peripheral nerves. The disease affects about one in 2,500 individuals worldwide, and there are no treatments for it. The researchers studied a form of the condition called Charcot-Marie-Tooth disease type 2A, which is caused by specific genetic mutations.
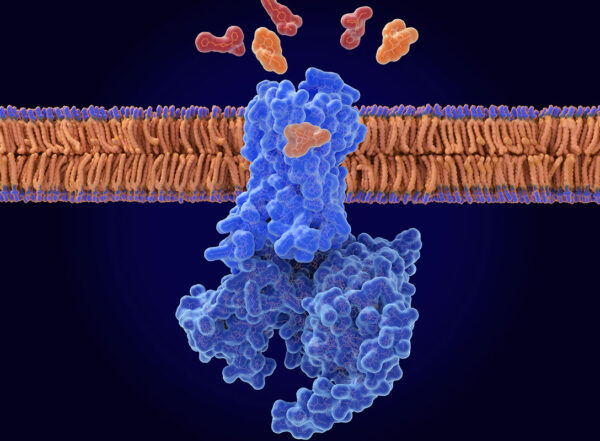
These patients have inherited mutations that affect mitochondria, the energy factories of cells. Healthy mitochondria fuse together and exchange mitochondrial DNA. This healthy mitochondrial “sex” is impaired in this disease because of mutations in a protein called mitofusin2, which governs mitochondrial fusion.
Because they can’t fuse, the mitochondria of people with this disease appear small, granular and clumped when viewed under a microscope. Until now, the small size was thought to be the main problem in the disorder. Small energy factories can’t produce enough fuel to keep nerves alive, so the cells slowly die off, the thinking went. But senior author Gerald W. Dorn II, MD, the Philip and Sima K. Needleman Professor of Medicine, suspected something else was going on.
Dorn knew the genetic mutations in this disease aren’t confined to nerves; rather, they are present in all of the mitochondria in every cell of the body. Nerve cells and, for example, heart muscle cells burn energy at high rates and need healthy, robust mitochondria for fuel. A cardiologist by training, Dorn wondered why patients with the disease don’t have heart problems.
“This disease starts with nerve loss in the feet, moves up the legs, then to the arms, but it doesn’t have major effects elsewhere,” said Dorn, also director of the School of Medicine’s Center for Pharmacogenomics. “People with Charcot-Marie-Tooth eventually may need wheelchairs, but they have normal lifespans.
“We found that the problem isn’t mitochondria that are too small, but mitochondria that can’t travel distances. In heart cells, mitochondria are packed like sardines and don’t need to move much, so there’s no problem with energy supply,” he said. “But for mitochondria to make it down a person’s leg — following the sciatic nerve from the lumbar spine to the foot — that’s akin to a 500-mile trip. If a person can’t constantly renew mitochondria, over the years the nerves start to atrophy. Once the nerves die, the muscles atrophy as well.”
With distance identified as a key factor, the ideal animal model to study this disease, Dorn quipped, is the giraffe. But since that’s not an option and mice don’t develop symptomatic Charcot-Marie-Tooth (because mice are small and their mitochondria don’t have far to go), Dorn and his colleagues extracted sciatic nerves — the longest nerve in the body — from mice and set up a way to compare the speeds of mitochondria moving up and down the nerves.
Co-first author Antonietta Franco, PhD, a postdoctoral researcher, found that mitochondria in the mouse nerves with Charcot-Marie-Tooth type 2A mutations were almost static, moving very little in the time frame observed, even when sped up with time-lapse photography. In contrast, mitochondria moving along the nerve axons of normal mice, viewed under the same time-lapse conditions, resembled an aerial shot of a highway.

The researchers then added a drug compound to the nerves with the static mitochondria. This drug, which they designed, works something like a chemical key that unlocks mitofusin2 and boosts mitochondrial fusion. The work developing the drug compound was led by co-first author Agostinho G. Rocha, PhD, a staff scientist.
“After about 15 minutes of exposure to the compound, the mitochondrial traffic began to pick up,” Dorn said. “And after an hour, it looked like the normal nerve. We also found that the drug doesn’t have an effect when added to normal nerves. Mitochondria appear to have a speed limit.”
While the researchers know this drug boosts the ability of diseased mitochondria to fuse to one another, Dorn said it’s possible this is not what lets them move. It may be that the unlocked form of mitofusin2 also lets mitochondria couple to what Dorn calls the railroad tracks of cells. But more research is needed to answer that question.
Dorn said these findings may be relevant for other neurodegenerative disorders in which mitochondria are impaired, including other types of Charcot-Marie-Tooth disease.
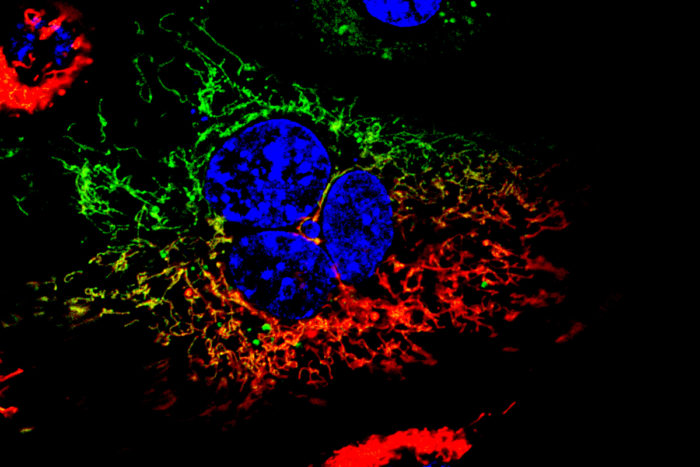 Antonietta Franco
Antonietta Franco